Değerli Metal Takıların Saflığını Test Etmek İçin Doğru Yöntemleri Keşfedin
Mücevherlerdeki Değerli Metal İçeriğini Test Etme Rehberi

İçindekiler
Bölüm Ⅰ Değerli Metal Takıların Saflığını Test Etme İlkeleri
Değerli metal takıların saflığını test etmek eski çağlardan beri var olmuştur. Atalarımız, değerli metal takıların saflığını belirlemek için testler yaparken öncelikle duyusal algılarına ve mevcut deneyimlerine güvenirlerdi. Örneğin, rengini gözleriyle gözlemler, elleriyle tartar ve ısırarak sertliğini test ederlerdi. Elbette, bunun arkasında belirli bir bilimsel gerekçe de vardır. Ancak, bilim ve teknolojinin gelişmesiyle birlikte, bilimsel test cihazlarının sürekli olarak icat edilmesi ve güncellenmesi, özellikle ticari testlerde, değerli metal takıların saflık testlerine bazı modern bilimsel test araçlarının eklenmesini sağlamıştır.
Değerli metal takıların saflığını test etmek için kullanılan modern test teknolojisi, doğruluk, kısa test süresi, düşük maliyet ve kullanım kolaylığı ile öne çıkan bilimsel cihazlara dayanmaktadır. Daha hızlı, daha basit ve daha doğru yöntemlere doğru gelişmektedir. Bilim ve teknolojinin sürekli gelişmesiyle birlikte, değerli metal takıların saflığını test etme teknikleri ve yöntemleri daha da gelişecektir.
Değerli metal takıların saflığının test edilmesinde genel olarak aşağıdaki üç ilkeye uyulması gerekir.
(1) Mümkün olduğunca tahribatsız muayene yapılmalıdır. Bu nedenle, test yöntemleri seçilirken değerli metal takıların görünümüne zarar vermeyen yöntemler tercih edilmelidir. Gerçekten kaçınılmazsa, danışanın onayı veya izni alınmalıdır.
(2) Tespit belirli bir doğruluk düzeyini korumalıdır. Başka bir deyişle, tespit doğruluğunun ilgili standart aralıkta olması gerekir.
(3) Tespit maliyeti mümkün olduğunca düşük olmalıdır.
Değerli metal takıların kalitesinin belirlenmesinde esas olarak iki hususa dikkat edilir: Birincisi, değerli metal takıların orijinalliğini belirlemek; ikincisi ise değerli metal takıların kalitesini belirlemektir.
Bölüm II Değerli Metal Takı Kalitesi için Yaygın Basit Tespit Yöntemleri
Antik çağlardan beri insanlar, değerli metallerin kalitesini ve orijinalliğini özelliklerine göre belirlemek için bir dizi deneysel yöntem kullanmışlardır. Bu yöntemlerin doğru bir şekilde kullanılması, değerli metal takıların orijinalliğini ve kalitesini etkili, hızlı ve kaliteli bir şekilde belirleyebilir.
1. Renk Gözlem Yöntemi
Eski insanlar, altının rengi ile içeriği arasında belirli bir benzerlik olduğunu fark etmişlerdi. Halk arasında şöyle bir söz vardır: "Dört yedi altın değildir." "Yedi yeşil", altın içeriğinin ve gümüş içeriğinin olduğu durumlarda altının yeşilimsi sarı göründüğünü ifade eder; "Sekiz sarı", altın içeriğinin ve gümüş içeriğinin olduğu durumlarda altının altın sarısı göründüğünü ifade eder; "Dokuz mor", altın içeriğinin ve gümüş içeriğinin olduğu durumlarda altının morumsu sarı göründüğünü ifade eder; "On kırmızı", altın içeriğinin 0'e yakın ve gümüş içeriğinin son derece düşük olduğu durumlarda kırmızı altın, saf altın veya saf altının kırmızımsı sarı göründüğünü ifade eder. Deneyimi özetlemenin bu geleneksel yöntemi, yalnızca gümüş içeren berrak altını değerlendirmek için etkilidir.
Saf altın, saf altın, kırmızı altın, saf kırmızı altın, 999 ayar altın ve 24 ayar altın için altın sarısının üzerinde hafif kırmızımsı bir ton bulunur. Halk arasında "kırmızı altın" veya "saf altın" olarak anılan renk, saf altının bu rengidir. K altın, 22 ayar, 18 ayar, 14 ayar, 10 ayar, 9 ayar, 8 ayar gibi altın takıların rengi, altındaki safsızlıkların türlerini ve oranlarını yansıtır. Genel olarak, gümüş içeren berrak altın serisinin rengi sarı, bakır içeren karışık altın serisinin rengi ise kırmızıdır.
Altının kalitesini, görünen rengine göre değerlendirmek ancak nitel bir tanımlama olabilir. Modern bilim ve teknolojinin gelişmesiyle, daha önce de belirttiğimiz gibi, farklı kalitedeki altınlar aynı rengi gösterebilir. Doğal altının kalitesini belirlemek için bu yöntemi kullanmanın bazı gerekçeleri vardır.
Geleneksel el işçiliği takılarında, sahte gümüş takılarda genellikle alüminyum veya alüminyum alaşımları, beyaz bakır, kalay veya kalay alaşımları kullanılır; bunlar genellikle donuk gri renge ve zayıf parlaklığa sahiptir; düşük gümüş içeren takılar hafif sarı veya gri renge ve zayıf rafineliğe sahiptir; yüksek gümüş içeren takılar parlak, saf beyazdır ve daha iyi parlaklığa sahiptir. Genel olarak konuşursak, takı gümüş ve bakır alaşımı olduğunda, 85 gümüş hafif kırmızımsı, 75 gümüş kırmızımsı sarı, 60 gümüş kırmızı ve 50 gümüş siyah görünür; takı gümüş ve beyaz bakır alaşımı olduğunda, 80 gümüş grimsi beyaz ve 50 gümüş siyah-gri görünür; takı gümüş ve pirinç alaşımı olduğunda, gümüş içeriği ne kadar düşükse, takı rengi o kadar sarıdır. Genellikle, saf beyaz ve ince işçilikle yapılmış takıların kalitesi 'ın üzerindedir. Buna karşılık, gri, kırmızı ve kaba işçilikli beyaz takıların kalitesi yaklaşık iken, gri-siyah veya açık sarı-kırmızı takıların kalitesi genellikle 'ın altındadır. Modern işçilikle üretilen imitasyon gümüş veya düşük gümüş içerikli takıların, yüzeyleri gümüş veya rodyumla kaplandığında, renk, hassasiyet ve yüzey parlaklığının gerçek gümüş takılardan ayırt edilemeyecek düzeyde olabileceği ve bu nedenle takıların kalitesini görsel olarak değerlendirmenin imkansız hale gelebileceği unutulmamalıdır.
Platinin kalitesi ve alaşım elementlerinin bileşimi farklılık gösterir ve bu da farklı renklerin görüntülenmesine neden olur: Daha kaliteli platin, hafif gri bir renge sahip mavimsi beyaz bir renk sunar. Belirli miktarda Cu veya Au içeren platin, hafif sarı bir renge sahip mavimsi beyaz bir renk sunar. Daha yüksek miktarda Ag içeren platin ise gümüş beyazı bir renk sunar. Paladyum takılar genellikle iyi bir metalik parlaklıkla çelik beyazı bir renk sunar. Taklit platin veya paladyum takılar genellikle oksidasyona ve matlaşmaya eğilimli beyaz bakır, nikel alaşımları, sodyum alaşımları vb. malzemelerden yapılır.
2. Touchstone Test Yöntemi
Dokunma taşı yöntemi, altın ve gümüşün gerçekliğini ve kalitesini belirlemek için kullanılan en eski araç ve yöntemdir ve dünya çapındaki eski medeniyetlerde kullanıldığına dair kayıtlar bulunmaktadır. Bu yöntem, test edilen mücevherlerin ve altın standardının (belirli bir kaliteye sahip, standart olarak adlandırılan bir dizi altın plaka) dokunma taşı üzerine çizilmesini içerir. Dokunma taşı üzerinde kalan çiziklerin rengi karşılaştırılarak mücevherin gerçekliği ve kalitesi belirlenebilir. Bu test yöntemi uzun zamandır nispeten doğru, güvenilir ve hızlı bir tespit yöntemi olarak kabul edilmektedir. Günümüzde bile birçok altın ve gümüş geri dönüşüm atölyesi, hem altın hem de gümüş takıların kalitesini tespit edebilen malzemeleri hızlı bir şekilde tanımlamak için bu yöntemi sıklıkla kullanmaktadır.
Geleneksel mihenk taşları çoğunlukla siyah veya gri taşlardır, genellikle siyah çakmaktaşı veya silisli arduvazdan yapılırlar, yaklaşık 6,5 Mohs sertliğine ve ince bir dokuya sahiptirler. Çin'in Sincan bölgesindeki antik altın ve bakır sahasının yakınındaki koyu silisli kaya çakılları, Gobi Çölü'nde "çöl cilası" olarak bilinen koyu silisli kayalar ve Nanjing'den gelen siyah yağmur çiçeği taşları, öğütüldükten sonra mükemmel mihenk taşlarına dönüştürülebilir. Altın test plakası, farklı standart saflıklara sahip altından yapılmış, bir ucuna altın plakanın standart saflığı kazınmış, genellikle birden fazla parçadan oluşan gruplara diş açmak için üzerinde küçük bir delik bulunan ince, küçük bir plakadır (Şekil 6-1'de gösterildiği gibi). Altın test plakası sınıflandırması ne kadar ince olursa, kapsanan renk aralığı o kadar geniş olur ve analiz sonuçları o kadar doğru olur.

Altın takıların saflığını test etmek için kullanılan ölçü taşı yöntemi, kolorimetrik bir yöntemdir. Yöntem şu şekildedir:
(1) Ölçü taşını hazırlayın.
Test taşının çalışma yüzeyini suyla yıkayın, durulayın ve kurulayın. Hint yağı ile kaplanmış taş yüzeyinde, test taşının uçlarına kadar uzanan bir yağ kanalı oluşturun, 20 mm genişlik uygundur, yağladıktan sonra temiz bir ipek bezle yüzeyde kalan yağı silin, böylece yağ kanalı çok ince bir tabaka halinde kalsın. Yağ tabakası çok kalın olduğunda, yağın akması ve siyahlaşması kolaydır, ancak çok kuru olduğunda renklenmesi kolay değildir. Yağ kanalının kenarı düz, aynı hizada, test taşının kenarına paralel olmalı ve altın kanalının aynı uzunlukta taşlanmasını sağlamak için belirgin bir ayrım oluşmamalıdır. Parmaklarınıza dikkat edin. Taş yüzeyine dokunmayın; özellikle ağızda gaz ve elde ter olan toz ve nemle lekelenmiş yüzeyden kaçının. Aksi takdirde renklendirmek emek gerektirir.
(2) Öğütme yöntemi.
Bir mihenk taşı bileme işlemi için kullanıldığında, genellikle taşı sol elinizle, altını ise sağ elinizle tutun; başparmağınız üstte, diğer parmaklarınız altta olsun. Yağlanmış taraf üstte olmalı ve mihenk taşı elinizde sıkıca tutulmalı ve hareket ettirilmeden masanın üzerinde sabitlenmelidir. Bileme sırasında, test edilecek nesne veya test plakası taş yüzeyine sıkıca bastırılmalı ve altını tutan sağ el bilek gücünü kullanmalıdır. Bileme yolu genellikle 20-30 mm uzunluğunda ve 3-5 mm genişliğindedir. Altın parçası yolu ve test plakası yolu uzunluk ve genişlik bakımından tutarlı olmalı ve renk karşılaştırması için test plakası yolu, altın parçası yolunun her iki tarafında bilenebilir. Altın parçası yolunun rengi test plakası yoluyla uyuşmuyorsa, bilemek için başka bir test plakası seçin ve iki yol eşleşene kadar rengi gözlemleyin.
(3) Kimlik
Altın, mihenk taşına çizildiğinde renkli bir iz bırakır. Uzun süreli uygulamalar sonucunda, insanlar mihenk taşı kullanarak altının gerçekliğini ve kalitesini belirlemek için "renk düzlemine bak, ışığa açılı bak, sesi dikkatlice dinle" olarak bilinen bir dizi deneyimi özetlemişlerdir. Gümüş içeren saf altın yumuşaktır ve altın yolu, dalgalı renk olmadan mavimsi görünür; esas olarak "renk düzlemine bak" ve ikincil olarak "yüzen renge açılı bak" üzerine odaklanır. Gümüş ve bakır içeren karışık altın için, taşlama sırasında ses ve dalgalı ışık vardır; esas olarak "yüzen renge açılı bak" ve ikincil olarak "renk düzlemine bak" üzerine odaklanır. Altın yolunu aşındırmak için asit kullanmak, renk farklılıklarını artırabilir ve ayırt edici özellikleri vurgulayabilir. Kullanılan asit, değerli metal malzemelerdeki temel metaller ve gümüşle tercihen reaksiyona girmelidir. Alaşım kalitesine bağlı olarak, kullanılan asitler nitrik asit, nitrik asit ve tuz karışımı veya nitrik asit ve hidroklorik asit karışımı vb. içerebilir.
Altın takıların kalitesini test etmek için kullanılan mihenk taşı yöntemi, görsel gözlem ve karşılaştırma yoluyla belirlenir, zengin pratik deneyim gerektirir ve birçok insan faktöründen etkilenir, bu da sınırlı bir doğruluk oranına yol açar. Ayrıca, altın takı çeşitleri arttıkça ve bileşimleri daha karmaşık hale geldikçe ve sınırlı sayıda altın kardeş kartıyla, altın kaplamalı veya altın kaplamalı ürünler arasında ayrım yapmak zorlaşmaktadır. Altın için tahribatsız muayene teknolojisinin sürekli gelişmesiyle, mihenk taşı yöntemi yavaş yavaş yerini daha kullanışlı, basit ve hassas yöntemlere bırakmıştır.
3. Tartım yöntemi
Altın yüksek bir yoğunluğa sahiptir, saf altının yoğunluğu 19,32 g/cm3'tür.3 Yoğunluk. Elle tartıldığında ağır hissedilir ve belirgin bir ağırlık hissi verir. Altının yoğunluğu kurşun, gümüş, bakır, kalay, demir ve çinko gibi metallerden çok daha fazla olduğundan, ister pirinç olsun (8,9 g/cm3 yoğunluğa sahip olsun) ister olmasın, altının yoğunluğuna bağlı olarak değişir.3), bakır bazlı alaşımlar veya nadir altın, alt altın, taklit altın vb. gibi imitasyon altın malzemeler veya altın kaplamalı ürünler, elle tartıldığında saf altının verdiği ağır hissi vermez. Tartım yöntemi, 24 ayar altını ayırt etmek için en etkili yöntemdir. Yine de, altına benzer bir yoğunluğa sahip tungsten alaşımından yapılmış altın kaplamalı veya altın kaplamalı ürünleri tespit etmek için daha etkili olabilir, çünkü ikisi arasındaki farkı elle hissetmek zordur.
Platinin yoğunluğu 21,45 g/cm3'tür3ve aynı hacimdeki platinin kütlesi gümüşün (10,49 g/cm3 yoğunluğunda) kütlesinin iki katından fazladır3). Altından daha yoğundur ve elle tartıldığında ağırlaşır. Bu nedenle, tartma yöntemiyle platin, altın ve gümüş takıları birbirinden ayırırken şöyle bir söz vardır: "Ağır olan platin veya altındır, hafif olan gümüş veya pirinçtir."
Gümüş ile alüminyum ve paslanmaz çelik arasında da önemli bir yoğunluk farkı bulunduğundan, tartım yöntemi kullanılarak da ayırt edilebilir: “Alüminyum hafiftir, gümüş ağırdır, bakır ve çelik ürünleri ne hafiftir ne de ağırdır.”
4. Süneklik Yöntemi
Mücevherlerin bükülme kolaylığı, dolaylı olarak altın mücevherlerin saflığını ve değerli metal malzemenin türünü de gösterebilir. Saf altın mükemmel esnekliğe sahiptir ve bu, altının yüksek tokluğunun ve düşük sertliğinin kapsamlı bir göstergesidir. Gümüş ikinci sıradadır, platin gümüşten daha serttir ve bakır en yüksek sertliğe sahiptir. Altın-gümüş alaşımları biraz daha serttir ve altın-bakır alaşımları daha da serttir; alaşımdaki altın içeriği ne kadar düşükse sertlik o kadar yüksektir. Örneğin, saf altın mücevherler ağızlarından veya tokalarından hafifçe büküldüğünde çok yumuşak hissedilirken, imitasyon altın malzemeler bu hissi vermez. Bu nedenle, saf altın kolayca bükülüp kırılırken, düşük saflıktaki altın mücevherler bükülmesi kolay değildir ve kırılmaya eğilimlidir.
Altın ve gümüş takıları test etmek için bu yöntemi kullanırken, takı genişliğinin ve kalınlığının esnekliği üzerindeki etkisine özellikle dikkat edilmelidir. Genellikle, daha geniş ve kalın takılar büküldüğünde daha sert hissedilir; tersine, daha dar ve ince takılar daha yumuşak hissedilir.
5. Sertlik Testi Yöntemi
Değerli metal takıların sertliği, altın içeriğiyle yakından ilişkilidir; saflık ne kadar yüksekse sertlik o kadar düşüktür. Saf altının sertliği çok düşüktür; yaygın bir yöntem dişlerle ısırmaktır. Dişlerin sertliği altından daha fazla olduğundan, altın üzerinde ısırık izleri kalabilir ve bu da yüksek saflıkta altın olduğunu gösterir. Buna karşılık, imitasyon altın malzemeler daha yüksek bir sertliğe sahiptir ve bu da ısırık izi bırakmayı zorlaştırır. Test sırasında, genellikle mücevherin arkasını veya göze çarpmayan bir alanını hafifçe çizmek için sert bir bakır iğne kullanılır; kalan çizik ne kadar derinse, altın içeriği o kadar yüksektir ve çizik belirgin veya yüzeysel değilse tam tersi geçerlidir. Ticari testlerde, değerli metal takıların saflığını test etmek için bu yöntemin kullanılmasının yıkıcı test olarak kabul edildiğini ve müşterinin onayı veya yetkilendirmesiyle yapılması gerektiğini unutmamak önemlidir.
Saf gümüşün sertliği de düşüktür ve tırnakla çizilebilir. Takı yumuşak ve sert değilse kalay veya kurşun içerebilir; sertse bakır (nikel gümüş), demir veya diğer alaşımlardan yapılmış olabilir.
6. Yangın Testi Yöntemi
Atasözünde de söylendiği gibi, "Gerçek altın ateşten korkmaz" ve "Yoğun ateş gerçek altını ortaya çıkarır." Altının yüksek bir erime noktası (1063°C) vardır ve yüksek sıcaklıklarda (erime noktasının altında) erimeden, oksitlenmeden ve rengi değişmeden kalabilir. Sıcaklık erime noktasını aşsa ve altın erimeye başlasa bile, rengini korur. Buna karşılık, düşük ayarlı altın ve taklit altın malzemeler, kırmızıya yakın bir sıcaklıkta yakılıp soğutulduğunda renk değiştirir, hatta siyaha döner.
Platinin erime noktası (1773°C), altından daha yüksektir. Yakılıp soğutulduktan sonra rengi değişmezken, gümüş, içindeki gümüş oranına bağlı olarak yakılıp soğutulduktan sonra süt beyazı, kırmızımsı veya siyahımsı kırmızıya döner.
7. Ses ve tonu dinleme yöntemi
Altın, gümüş ve platinin sertliği düşük olduğundan, katı altın veya yüksek ayarlı altın takılar havaya atıldığında, yere düştüğünde çıkardığı ses donuktur, gürültü veya sekme olmaz. Takı sert bir çimento zemine düştüğünde, yüksek ayarlı altın veya platin takılar az esnekliğe sahip donuk bir ses çıkarır; düşük ayarlı takılar, bakır veya paslanmaz çelik ürünler yüksek sekmeyle keskin ve yüksek bir ses çıkarır. Geleneksel saf altının sesi vardır ancak tonu yoktur ve çok az sekme vardır, karışık altının ise sesi, tonu ve sekmesi vardır; daha fazla sekme, daha keskin ve daha uzun tonlar daha düşük saflığı gösterir. Bununla birlikte, altın takı üretim teknolojisindeki gelişmelerle birlikte, mevcut pazarda 999 altın standartlarını karşılayan ve iyi esnekliğe sahip birçok yüksek mukavemetli sertleştirilmiş katı altın ürünü ortaya çıkmıştır.
Platinin yoğunluğu altından daha fazladır ve havaya atıldığında ve yere düştüğünde çıkardığı ses özellikleri altınınkine benzemektedir, bu da imitasyon platin, platin kaplamalı ve platin kaplamalı takıları ayırt etmede kullanılabilir.
Benzer şekilde, saf gümüş ve yüksek saflıktaki gümüş takılar yüksek yoğunluğa ve yumuşak bir dokuya sahiptir ve bu da bir yüzeye düşürüldüğünde düşük bir geri tepme yüksekliğine neden olur. Buna karşılık, sahte gümüş veya düşük saflıktaki gümüş takılar, düşük yoğunlukları ve sertlikleri nedeniyle nispeten daha yüksek bir geri tepme yüksekliğine sahiptir.
8. İşaretleme yöntemi
Altın takılar, saflığını belirtmek için uluslararası standartlara uygun olarak damgalanmalıdır. Ülkemizde 24 ayar altın, "saf", "saf altın", "kırmızı altın" veya "24 ayar", 18 ayar altın ise "18 ayar" veya "750" gibi etiketlerle işaretlenir.
Ülkemizde gümüşün saflığı, binde bir, yüzde veya kesir ile ve ardından "s" (gümüş) harfi ile gösterilir; örneğin "800'ler", "80'ler" ve "80% S" gibi; bunların hepsi gümüşün saflığını gösterir; uluslararası alanda ise genellikle binde bir, ardından "S" veya "Gümüş" harfi ile gösterilir; örneğin "800 S" ve "800 Gümüş" gibi; bunların her ikisi de gümüşün saflığını gösterir. Ayrıca, uluslararası alanda genellikle "SF" (gümüş dolgunun baş harfleri) ile gösterilen gümüş kaplamalı bir malzeme mührü de vardır.
Uluslararası alanda platin saflığı ve kalitesi binde birlik sayının ardından gelen “Pt”, “Plat” veya “Platinum” ile belirtilir; örneğin 950Pt, platinin saflığını gösterir; Amerika Birleşik Devletleri'nde ise yalnızca platinin 'in üzerinde olduğunu garanti eden “Pt” veya “Plat” ile işaretlenir.
Bölüm III Hidrostatik Yöntem (Yoğunluk Yöntemi)
1. Algılama İlkesi
Saf altının yoğunluğu 19,32 g/cm3'tür3Belirli bir değerli metal süs eşyasının yoğunluğunun bu değerden düşük olması, içine başka metallerin karıştığını doğrulayabilir. Yoğunluğun boyutu, altının saflığıyla yakından ilişkilidir. Altının saflığı, yoğunluk yönteminin değerli metal takıların saflığını test etmek için kullanılmasının temel prensibi olan yoğunluktan çıkarılabilir.
Mücevherin hacmi, mücevherdeki saf altın hacmi ile saf olmayan metallerin hacminin toplamına eşittir, yani:
V = Vsaf + Vsafsızlıklar (6-1)
Formülde:
Aksesuarın V hacmi (mL);
Vsaf-Mücevherdeki saf altının hacmi (mL) kaçtır?
Vsafsızlıklar -Mücevherdeki safsızlıkların hacmi (mL)
Doğru tartım için 1/10000'lik bir analitik terazi kullanılarak altın mücevherin kütlesi m' olarak bulunur; ardından mücevheri sabitlemek için ince bir ip kullanılarak sudaki kütlesi m' olarak doğru bir şekilde ölçülür (gerekirse ipin kütlesi düşülmelidir). Arşimet ilkesine göre, sudaki bir cisme etki eden kaldırma kuvveti, yerinden oynattığı suyun kütlesine eşittir, yani:
m – m' = V x ρ su (6-2)
Suyun normal yoğunluğu 1 g/cm3'tür3 , şu sonucu verir: m – m' = V, Denklem (6-1) yerine konulduğunda şu elde edilir:
m – m' = Vsaf + V safsızlıklar
Cismin hacmi ile kütlesi arasındaki ilişkiye göre V = m/ρ şu şekildedir:

Yukarıdaki denklemi basitleştirip saf altın yoğunluğunu ρ saf = 19,32 g/cm3 olarak değiştirirsek3 Bunu kütle kesrine dönüştürürsek şunu elde ederiz:
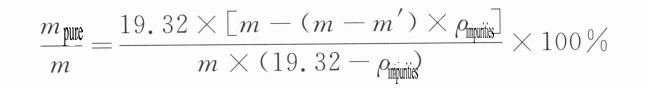
Formülde:
m – mücevher kalitesi (g);
m' – mücevherin sudaki kalitesi (g);
m saf – takıdaki saf altının kalitesi (g);
ρ safsızlıklar – mücevherdeki safsızlıkların yoğunluğu (g/cm3)3)
2. ρ safsızlıklarının değerinin alınması yöntemi
Yukarıdaki formül, altın takılardaki altın içeriğini tespit etmek için kullanılır ve analitik terazinin gerçek tartımıyla elde edilir. Safsızlıkların değeri henüz belirlenmemiştir. Altın takılardaki ana safsızlıklar olan Ag ve Cu'ya ilişkin deneyimlere göre, safsızlık yoğunluğu Ag ve Cu'daki safsızlıkların bağıl içeriğine göre belirlenir. Bunlar arasında Ag'nin yoğunluğu 10,49 g/cm3'tür.3ve Cu'nun yoğunluğu 8,90 g/cm3'tür3, bu nedenle safsızlıkların değeri 8,90 ~ 10,49/cm arasında değişmektedir3Kirlilik değerleri şu şekildedir:
Altın-gümüş serisi alaşımlar (berrak altın) için: ρ safsızlıklar = ρ Gümüş = 10,49 g/cm33
Altın-bakır serisi alaşımlar (karışık altın) için: ρ safsızlıklar = ρ bakır = 8,90 g/cm33
Altın – gümüş – bakır serisi alaşımlar (karışık altın) için: ρ safsızlıklar =1/(x/ρ Gümüş + y/ρ bakır), x+y = 1
Eğer x = y = 0,5 ise, o zaman ρ safsızlıklar =9,63 g/cm33
Eğer x:y = 1 : 2 ise, x = 0,3333, y = 0,6666, ρ safsızlıklar = 9,375
Eğer x:y = 2 : 1 ise, o zaman x = 0,6666, y = 0,3333, ρ safsızlıklar = 9.901
Yukarıdaki analiz, altın alaşımının yoğunluğunun ve farklı tür ve oranlardaki safsızlık metallerinin yoğunluğunun, altın takıların kalitesini doğru bir şekilde hesaplamak için temel faktörler olduğunu göstermektedir. Ancak test edilen numunedeki safsızlık metallerinin türleri ve oranları önceden bilindiğinde, numunenin kalitesi yoğunluk yöntemi kullanılarak hesaplanabilir; bu aynı zamanda yoğunluk testi için de gerekli bir koşuldur.
Hidrostatik yöntemin saf altın takılardaki altın içeriğini daha doğru bir şekilde belirleyebildiği unutulmamalıdır. Alternatif olarak, alaşım bileşenlerinin element oranları bilindiğinde, takıdaki altın içeriği, takıdaki tespit edilen yoğunluk değerine göre hesaplanabilir. Ancak, alaşımın bileşen oranları bilinmediğinde, takıdaki altın içeriğini tespit edilen yoğunluk değerine göre hesaplamak genellikle imkansızdır. Bu nedenle, alaşım bileşenleri belirsiz olduğunda, takıdaki altın içeriği ile yoğunluk değeri arasında birebir bir ilişki yoktur.
3. Yoğunluk Yöntemi Tespitinin Özellikleri
Yoğunluk yöntemi, Arşimet prensibini kullanarak mücevherlerin yoğunluğunu test eder ve altın-gümüş-bakır alaşımlarının yoğunluğuna bağlı olarak kalite içeriğini hesaplar. Bu yöntemin kullanım kolaylığı, hız, tahribatsız örnekleme, minimum ekipman ve kullanım kolaylığı gibi avantajları vardır. Altın takıların gerçekliğini, altın mı yoksa altın kaplama mı olduğunu belirlemek ve saf altın takıların altın içeriğini ölçmek gibi etkili bir şekilde ayırt eder. Yüzükler ve kırbaç zincirleri gibi dikişsiz damgalı takılar için muayene doğruluğu nispeten yüksektir. Ancak içi boş takıları test edemez. 19,35 g/cm3 yoğunluğa sahip tungsten gibi yüksek yoğunluklu safsızlıkları ayırt edemez.3Saf altına çok yakın olduğundan, bu yöntemle ölçüm yapmak zordur. K altın takıların kalitesini test etmedeki hata, özellikle takıların içinde kum delikleri ve kaynak delikleri, çalışma sıvısının nüfuz edemeyeceği yüzey boşlukları veya altın ve gümüş dışındaki safsızlıklar varsa önemlidir ve bu da tespit sonuçlarında hatalara yol açabilir.
4. Tespit Yöntemleri
4.1 Çift Pan Denge Yöntemi
4.1.1 Test Cihazları
0,1 mg hassasiyetli bir terazi, daldırma sıvısı, küçük bir masa ve ince bakır tel (yerine saç da kullanılabilir).
(1) Denge. 0.1mg hassasiyete sahip mekanik veya elektronik teraziyi tercih edebilirsiniz.
(2) Daldırma sıvısı. 50 mL'lik cam beher içerisinde bulunan susuz etanol, karbon tetraklorür, ksilen, su veya su ile karıştırılmış etanolü seçebilirsiniz.
(3) Küçük masa. Terazi modeline göre metal bir levhadan yapılmış küçük bir masa, tartım kefesinin yukarı-aşağı hareketini etkilemeyecek şekilde tartım kefesinin üzerine yerleştirilebilir.
(4) İnce bakır tel. Eşit uzunlukta (Φ=0,2 mm) ince bakır telden birkaç parça kesin, bunları teraziyle tartın ve her gruptan eşit toplam kütleye sahip iki segment seçip iki gruba ayırın. Bir grubun iki küçük segmentinin bir ucunu küçük kancalara yuvarlayın ve diğer uçlarını birbirine bükün, böylece her iki küçük kanca da aynı anda tartım kefesine asılabilir [Şekil 6-2(a)], bir uç numune tutucuya takılabilirken diğer uç çözeltiye daldırılabilir [Şekil 6-2(b)]; diğer grup doğrudan ağırlık kefesine yerleştirilebilir. Saç kullanılıyorsa, ince bakır telin tüm ayrıntıları ve işlem adımları atlanabilir ve saçı altın takıya bağlayarak küçük bir halka yapılabilir ve numune tutucunun orta kancasına asılabilir.
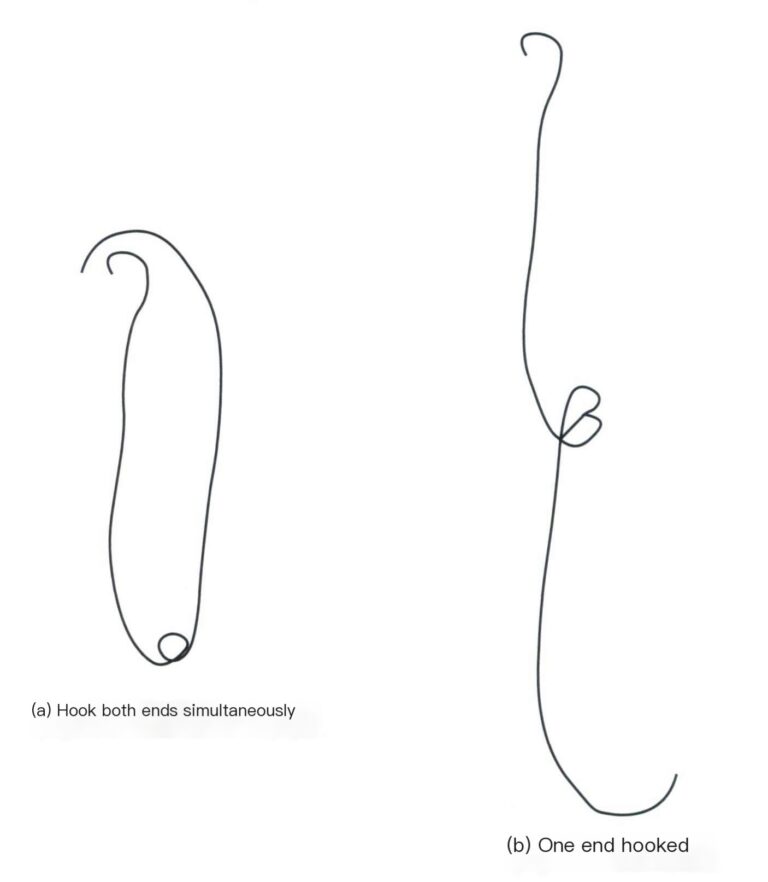
4.1.2 Çalıştırma Adımları
(1) Sıfır noktalarının dengesini kontrol edin. İnce bakır teli çıkarın, denge sıfır noktasını belirleyin, hassasiyet 0,0001 g'dan büyük olmayacak şekilde vidayı ayarlayın, ardından ince bakır teli her iki tarafa asın ve işaretçi "0" konumuna hizalanacak şekilde denge sıfır noktasını ayarlayın; saç kullanıyorsanız, bakır teli astıktan sonra denge sıfır noktasını ayarlama adımı atlanabilir.
(2) Sıcaklık düzeltme eğrisini belirleyin. Daldırma çözeltisinin yoğunluğu farklı sıcaklıklarda değişir. Tablo 6-1, etanol, ksilen ve karbon tetraklorürün farklı sıcaklıklardaki yoğunluklarını listelemektedir. Uygulamada, organik çözeltinin saflığı, sonradan eklenen safsızlıklar ve termometre ile daldırma çözeltisi beherinin sıcaklığı arasındaki sıcaklık farkı, ölçülen sonuçların Tablo 6-1'deki verilerden sapmasına, hatta bazılarının önemli ölçüde farklı olmasına neden olabilir.
Tablo 6-1 Farklı Sıcaklıklarda Etanol, Ksilen ve Karbon Tetraklorür Daldırma Çözeltilerinin Yoğunlukları
| İnfüzyon | |||||
|---|---|---|---|---|---|
| Etanol | Etanol | Ksilen | Ksilen | Karbon tetraklorür | Karbon tetraklorür |
| Yoğunluk /(g/cm3) | Sıcaklık /℃ | Yoğunluk / (g/cm3) | Sıcaklık /℃ | Yoğunluk /(g/cm3) | Sıcaklık /℃ |
| 0.837 | 7 | 0.839 | 6 | 1.630 | 3 |
| 0.830 | 16 | 0.829 | 16 | 1.610 | 13 |
| 0.829 | 18 | 0.824 | 22 | 1.599 | 18 |
| 0.827 | 19 | 0.819 | 27 | 1.589 | 23 |
| 0.821 | 21 | 0.814 | 32 | 1.579 | 28 |
| 0.817 | 26 | 0.809 | 37 | 1.569 | 33 |
| 0.810 | 32 | 0.804 | 42 | 1.559 | 38 |
(3) Değerli metal takıları iyice temizleyin ve kuruyana kadar susuz etanol veya asetonla silin.
(4) Altın takıları tartı kabının orta kancasına ince bakır tel veya saçla asın ve değerli metal takıların kütlesini tartın.
(5) Değerli metal takıları daldırma sıvısı kabına daldırın ve daldırma sıvısı içindeki altın takıların kütlesini m tartın.
(6) Değerli metal mücevherin yoğunluğunu hesaplayın ρ altın =m/(m-m') x, daldırma sıvısının yoğunluğuna göre.
(7) Yoğunluğa ve varsayılan son üye metallere dayanarak değerli metalin (altın veya gümüş) saflığına dönüştürün.
4.1.3 Notlar
(1) Değerli metal takılar temiz ve kuru olmalıdır; aksi takdirde hata önemli olacaktır.
(2) Çalışma eğrisi düzenli olarak kalibre edilmelidir; bir kerelik bir düzeltme olamaz.
(3) Değerli metal takılar çözeltiye daldırıldığında, hemen tartmayın; bir süre çalkalayın ve kabarcık olup olmadığını görsel olarak kontrol edin. Görünürde küçük kabarcıklar varsa, bunlar giderilmelidir.
(4) Etanol, ksilen ve karbon tetraklorür uçucu maddelerdir; ölçümler hızlı ve kararlı olmalı ve teraziye dökülmemesine dikkat edilmelidir. Ölçümden sonra, özel bir kapakla örtün veya özel bir şişeye boşaltın; orijinal kabına geri dökmeyin.
(5) Yoğunluğun altın yoğunluğunu aştığı bir durum varsa kalibrasyon yapılmalıdır.
(6) Değerli metal takıların adı, kalitesi, şekli, yüzey yapısı ve rengi, özellikle de renk ve yüzey özellikleri kaydedilmelidir. Yapı, tungsten içeren takıların kalitesindeki farklılıkları önleyebileceği için çok önemlidir. Orijinal verilerin saklanması, tespit hatalarının analizine olanak tanır ve bu da kalite yönetimi açısından faydalıdır.
4.2 Tek tavalı elektronik terazi yöntemi
4.2.1 Enstrüman
0,0001g hassasiyetli elektronik tek kefeli terazi, daldırma sıvısı ve süspansiyon rafı.
(1) Elektronik terazi. Tek tava, 0.0001g veya daha fazla hassasiyet, dijital ekran.
(2) Daldırma sıvısıÇift tava yöntemine benzer şekilde, denge askısı olmadığından, tutmak için biraz daha büyük bir kap kullanılabilir.
(3) Süspansiyon çerçevesi. Tartım kefesinin yukarı aşağı hareketini etkilemeyecek şekilde tartım kefesinin dışına sabitlenerek daha büyük yapılabilir, daldırma sıvısı beherinin 1,5 ~ 2 katı yükseklikte olabilir; ayrıca tartım kefesine yerleştirilerek, daldırma sıvısının içinde elle tutularak veya terazi kapağına kanca yapılarak numunenin terazi kapağına asılmasıyla havada tartım yapmak da mümkündür.
4.2.2 Çalıştırma adımları
(1) Terazinin sıfır noktasını kontrol edin; inceleme için elektronik terazinin kullanım kılavuzuna bakın.
(2) Çift tava yöntemini kullanarak sıcaklık düzeltme eğrisini belirleyin.
(3) Değerli metal takıları çift tava yöntemini kullanarak yıkayın ve kurulayın.
(4) Daldırma sıvısı kabını tartım tepsisine yerleştirin, süspansiyon çerçevesini takın, daldırma sıvısını dökün ve teraziyi sıfıra ayarlayın.
(5) Değerli metal takıyı tartım tepsisine yerleştirin, değerli metal takı m'nin kütlesini okuyun ve kaydedin.
(6) Değerli metal takıyı saçla süspansiyon çerçevesine asın, daldırma sıvısına batırın, değerli metal takıların havadaki ve daldırma sıvısındaki kütle farkını (m-m') doğrudan okuyun ve kaydedin.
(7) Çift tava yöntemini kullanarak değerli metal takıların yoğunluğunu hesaplayın.
(8) Değerli metal takıların inceliğini çift tava yöntemini kullanarak dönüştürün.
4.2.3 Notlar
(1) Tek tava yönteminde masa tablası yoktur ve daldırma sıvısının uçuculuğu doğruluğu önemli ölçüde etkiler. Bu nedenle, kütlenin sıfırlanması ile ölçülmesi arasındaki süre kısa olmalı ve ölçümler, özellikle iki ölçüm arasındaki zaman aralığının en aza indirilmesi gereken yaz aylarında, hızlı ve istikrarlı olmalıdır.
(2) Numune kabı merkeze yerleştirilmeli ve daldırma sıvısı kabı merkeze yerleştirilmelidir; aksi takdirde ölçüm sonuçları etkilenecektir.
(3) Elektronik terazinin hassasiyeti kontrol edilmeli, ayrıca dijital gösterge sistemi de bilinen standartlara göre doğrulanmalıdır.
(4) Sıvıyı dökerken dikkatli olun ve sıvıyı elektronik terazinin yüzeyine dökmeyin.
Bölüm IV X-ışını Floresan Analiz Yöntemi (XRF Yöntemi)
X-ışını floresan spektroskopisi (XRF), metalurji, madencilik, petrol, çevre koruma, tıp, jeoloji, arkeoloji, suç soruşturması, tahıl ve petrol, finans ve diğer sektörlerde yaygın olarak kullanılan etkili bir analitik yöntemdir. Değerli metaller için X-ışını floresan spektroskopisi yöntemi, uluslararası finans kuruluşları tarafından önerilen test yöntemlerinden biridir.
1. X-ışını Floresan Analizinin Temel Prensipleri
Elektron probu, numune uyarıldıktan sonra yayılan karakteristik X-ışını spektral çizgilerinin dalga boyunu (veya enerjisini) ve yoğunluğunu belirler. X-ışını floresan analizi de buna benzer, ancak elektron probundan farklı olarak, gelen ışık X ışınlarıdır. Işınlanan numune birincil X-ışınlarını emer ve uyarılarak ikincil X-ışınları yayar. Çeşitli ikincil X-ışınları, X-ışını floresansı olarak adlandırılır ve bu karakteristik spektral çizgilerin dalga boyu (veya enerjisi) ve yoğunluğu ölçülerek element içeriği belirlenebilir.
2. X-ışını floresan spektrometresinin yapısı
1948'de Friedman (H. Friedman) ve Birks (LS Birks), dünyanın ilk ticari X-ışını floresan spektrometresini geliştirdiler. X-ışını floresan spektrometresi teknolojisi onlarca yıldır hızla gelişti ve hız, esneklik ve hassasiyetle karakterize edilen yeni modeller sürekli olarak ortaya çıktı. X-ışını floresan spektrometreleri iki ana kategoriye ayrılır: dalga boyu dağılımlı X-ışını floresan spektrometreleri ve enerji dağılımlı X-ışını floresan spektrometreleri. Bunlardan ilki, ardışık ve eş zamanlı olmak üzere iki kategoriye daha ayrılabilir.
2.1 Sıralı dalga boyu dağılımlı X-ışını floresan spektrometresi
Sıralı dalga boyu dağılımlı X-ışını floresan spektrometresi, temel olarak bir X-ışını tüpü, bir spektroskopik sistem, bir algılama sistemi ve bir kayıt sisteminden oluşur. Cihazın yapısı Şekil 6-3'te gösterilmektedir.
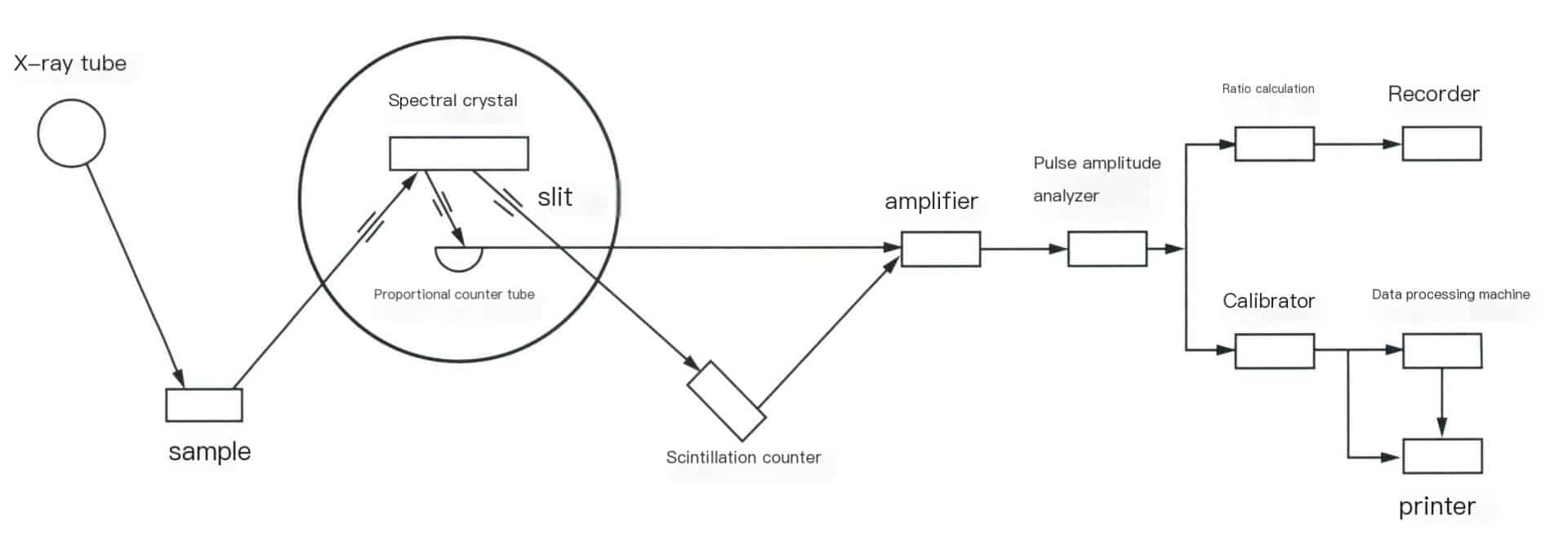
(1) X-ışını tüpü.
X-ışını tüpü, esasen elektron yayan bir katot ve elektron alan bir anot (hedef) içeren yüksek voltajlı bir vakum diyotu olan X-ışınları üreten cihazdır. Elektronlar, X-ışını tüpünün penceresinden yayılan ve numuneyi ışınlayan X-ışınları üretmek için anot hedef yüzeyini bombardıman eder. Pencere tarafından çeşitli dalga boylarındaki X-ışınlarının emilimini azaltmak için hafif eleman malzemeleri seçilir ve yaygın olarak kullanılan X-ışını tüplerinde genellikle berilyum pencereler kullanılır.
(2) Spektroskopik sistem.
Numune odası, yarıklar ve spektroskopik kristaller dahil olmak üzere çeşitli parçalardan oluşur. Numune odası, numune tepsileri, kutular, numune tutucular ve numune döndürme mekanizmaları gibi bileşenleri içeren numunelerin depolandığı yerdir. Numuneler katı (bloklar, plakalar, çubuklar, tozlar vb.) veya sıvı olabilir. Kolimatör veya prizma ızgarası olarak da bilinen yarık, numuneden üretilen ıraksak X ışınlarını keserek bunları spektroskopik kristal veya dedektör penceresine yansıtılan paralel ışınlara dönüştürmeye yarar. Spektroskopik kristalin rolü, farklı dalga boylarındaki spektral çizgileri ayırmak veya dağıtmaktır. Dispersiyonun temel prensibi, kristalin kırınım fenomenini kullanarak farklı dalga boylarındaki karakteristik spektral çizgileri ayırmak ve böylece ölçülen elementlerin karakteristik X ışınlarının belirlenmesi için seçilmesine olanak sağlamaktır.
(3) Algılama sistemi.
X-ışınlarını alır ve bunları ölçülebilir veya gözlemlenebilir sinyallere dönüştürür. Görünür ışık, elektrik darbe sinyalleri vb. gibi sinyaller elektronik devreler aracılığıyla ölçülür. Modern X-ışını floresan spektrometrelerindeki yaygın dedektörler arasında sintilasyon sayaçları, orantılı sayaçlar ve yarı iletken dedektörler bulunur.
Sintilasyon sayacı: Kısa dalga X-ışınları için yüksek algılama verimliliğine sahip, yaygın olarak kullanılan bir sayaçtır ve daha ağır elementler için algılama verimliliği, dalga boyu 3A'dan küçük X-ışınlarınınkine yakın olabilir. Bir sintilatör, fotoçoğaltıcı tüp, yüksek voltajlı güç kaynağı ve diğer bileşenlerden oluşur. X-ışını analizinde, ağır elementler için -, daha hafif elementler için ise - enerji çözünürlüğü sağlar.
Oransal sayaçlar: Kapalı oransal sayaçlar ve gaz akışlı oransal sayaçlar olarak ikiye ayrılır.
Orantılı sayaçlar, 3A'dan büyük dalga boylarına sahip X ışınlarını tespit etmek için kullanılır. Modern X-ışını spektrometreleri genellikle gaz akışlı orantılı sayaçlar kullanır. Uzun dalga X ışınlarının emilimini azaltmak için, dedektör pencere malzemesi olarak kullanılan alüminyum kaplı polyester film çok incedir (genellikle 6 µm, ancak daha ince olanları da mevcuttur). İnce pencere gaz sızıntısını engelleyemez, bu nedenle gaz akışı kullanılarak havayı dışarı atmak için taze gaz verilir. P10 gazı (90% argon, 10% metan) en yaygın kullanılan karışık gazdır. Orantılı sayaçların enerji çözünürlüğü, sintilasyon sayaçlarından daha iyidir.
Kapalı orantılı sayaçlar, gaz sızıntısını önlemek için inert gazlar, oksijen, azot vb. gibi iyonize gazları kalıcı olarak kapatır ve genellikle 12-15 µm kalınlığındaki nispeten kalın berilyum veya mika pencerelerle donatılmıştır. Diğer koşullar, gaz akışlı orantılı sayaçlarla aynıdır.
Yarı iletken dedektörler: Başlıca enerji dağılımlı spektrometrelerde kullanılırlar, yüksek algılama verimliliği ve enerji çözünürlüğü gibi avantajları vardır, hafif ve ağır elementlerin en karakteristik spektrumlarından enerji algılamasına olanak verirler.
(4) Kayıt sistemi.
Bir amplifikatör, darbe genliği analizörü ve okuma bölümünden oluşur. Amplifikatör: Bir ön amplifikatör ve bir doğrusal amplifikatör (ana amplifikatör) içerir. Sintilasyon sayaçları ve orantılı sayaçlardan gelen darbe genliği çıkışı genellikle onlarca ila yüzlerce milivolt arasında değişir; zayıf elektrik sinyalleri doğrudan sayılamaz ve yükseltilmelidir. Ön amplifikatör önce, genellikle on ila birkaç on kat arasında yükseltir ve ana amplifikatör giriş sinyali darbelerini daha da yükseltir. Bu da, sonraki ayrım devresinin gereksinimlerini karşılayan ve 500-1000 kata ulaşan yükseltme faktörleriyle darbe genlikleri elde edilmesini sağlar. Darbe genliği analizörü: İşlevi, belirli bir darbe genliği aralığı seçerek analiz hattının darbelerinin parazit ve arka plandan ayırt edilmesini sağlarken, paraziti bastırır ve analizin hassasiyetini ve doğruluğunu artırmak için maliyetleri düşürür. Okuma bölümü bir kalibratör, oran ölçer, yazıcı ve diğer bileşenlerden oluşur.
2.2 Eşzamanlı otomatik X-ışını floresan spektrometresi (çok kanallı X-ışını floresan spektrometresi olarak da bilinir)
Her biri kendi kristali, kolimatörü, dedektörü, amplifikatörü, darbe yüksekliği analizörü ve sayma kalibratörüne sahip, ortak bir X-ışını tüpü ve numunesi etrafında radyal olarak düzenlenmiş bir dizi tek kanallı cihazdan oluşur. Kanalların çoğu sabittir; yani belirli bir elementin spektral çizgilerini 20° açıyla analiz ederler ve o elementin spektral çizgisine en uygun bileşenlerle donatılmıştır. Bu tür kanallara sabit kanal denir. Şu anda 22 kanallı, 28 kanallı, 30 kanallı vb. cihaz modelleri görülebilir. Diğer bir kanal türü ise tarama kanalıdır; çok kanallı bir spektrometre, 2ϴ taramalı nitel analiz için motor tahrikli mekanizmalara sahip 1-3 tarama kanalına sahiptir.
Çok kanallı cihazlar, bir numunedeki çeşitli elementleri aynı anda belirleyebildiğinden, çok sayıda benzer numunenin analizi için uygundur. Ancak, bu tür cihazlar büyük bir yapıya sahiptir, pahalıdır ve uygulama alanları daha geniş olabilir.
2.3 Enerji dağılımlı X-ışını floresan spektrometresi
Dalga boyu dağılımlı X-ışını floresan spektrometresi ile enerji dağılımlı X-ışını floresan spektrometresi arasındaki karşılaştırma, yalnızca numuneden yayılan karakteristik X-ışınlarının ayrıştırılması (dağıtılması) konusundaki farktan kaynaklanmaktadır. İlki spektroskopi için kristaller kullanırken, ikincisi genellikle yüksek enerji çözünürlüğüne sahip bir yarı iletken dedektör ve enerji tarama analizi için çok kanallı bir darbe genliği analizörü kullanır. Modern bir enerji dağılımlı X spektrometresinin yapısı Şekil 6-4'te gösterilmektedir.
Enerji dağılımlı X-ışını floresan spektrometrelerinde, X-ışını kaynağı bir X-ışını tüpü veya uyarma kaynağı olarak radyoaktif bir izotop olabilir. Numune tarafından yayılan karakteristik X-ışınları, deteksiyon için bir yarı iletken dedektöre [yaygın olarak kullanılan Si(Li) dedektörü] gönderilir ve bu da genlik ve foton enerjisiyle orantılı bir dizi akım darbesi ile sonuçlanır. Dedektörün çıkışı yükseltildikten sonra, darbe analizi için çok kanallı bir darbe yüksekliği analizörüne gönderilir. Elde edilen çeşitli darbe yüksekliği dağılımları, enerji spektrumları olarak görüntülenir veya kaydedilir; burada görüntülenen görüntü, yoğunluk-darbe yüksekliği veya yoğunluk-foton enerjisi spektrumudur. Elementlerin konsantrasyonu (içeriği), enerji spektrumu tepelerinin yüksekliğine göre belirlenir.
Çoğu durumda, uyarma kaynağı olarak radyoaktif izotoplar kullanıldığından, bu tür X-ışınları "yumuşak" XX-ışınları olarak da bilinir. "Yumuşak" X-ışınlarıyla üretilen enerji dağılımlı X-ışını floresan spektrometresi, X-ışını kaynağıyla ilgili birçok bileşen ve sistemi ortadan kaldırdığı için hafiftir.
3. X-ışını floresan spektroskopik analizinin özellikleri
3.1 Avantajları
(1) Çok çeşitli elementler analiz edilebilir; periyodik tablodaki ilk 92 elementin neredeyse tamamı analiz edilebilir.
(2) Analiz edilebilen element içeriğinin aralığı oldukça geniştir, birkaç yüz binde birinden 0'e kadar değişir ve hassasiyet diğer tespit yöntemleriyle karşılaştırılabilir.
(3) Bu yöntem, tahribatsız bir analitik yöntemdir; yani numune analiz sürecinde hasar görmez, kimyasal durumunda değişiklik olmaz ve numunenin dağılmasına neden olmaz. Aynı numune tekrar tekrar ölçülebilir ve değerli metal takıların tespit ihtiyaçlarını karşılar. Özellikle değerli metal ürünlerinin kalite değerlendirmesi ve özgünlüğünün doğrulanması için uygundur.
(4) Analiz hızı yüksektir. Ölçüm için gereken süre, ölçümün hassasiyetine bağlıdır, ancak genellikle çok kısadır ve numunede ölçülecek tüm elementler 2-5 dakika içinde tamamlanabilir.
(5) Analitik numunenin morfolojisinden ve kimyasal bağlanma durumundan bağımsızdır; katı numuneler, sıvılar, preslenmiş bloklar, tozlar, filmler veya herhangi bir boyuttaki numuneler analiz edilebilir.
(6) Analiz maliyeti düşüktür ve operatörlerden beklenen mesleki altyapı ve teknik yeterlilik yüksek değildir.
3.2 Sınırlamalar
(1) Metalik olmayan elementler ve metaller ile ametaller arasındaki elementlerin doğru bir şekilde tespit edilmesi zordur. Temel parametre yöntemleriyle test yapılırken, test numunesi C, H veya O gibi hafif elementler içeriyorsa hatalar meydana gelir.
(2) Standart eğriler oluşturmak için temsili numunelere ihtiyaç vardır ve analiz sonuçlarının doğruluğu, diğer elementlerin girişimlerinden ve örtüşen tepe noktalarından kolayca etkilenebilen standart numunelerin kimyasal analizine dayanmaktadır. Standart eğri modelinin zaman zaman güncellenmesi gerekir; cihazda veya standart numunelerde değişiklik olduğunda, standart eğri modelinin de değişmesi gerekir.
(3) Radyoaktif izotop kaynaklarından kaynaklanan potansiyel bir kirlenme tehdidi bulunmaktadır.
(4) XRF yöntemi, farklı matrislere sahip altın takılar için büyük bir tespit hatasına sahiptir, numunelerin özelliklerini ve homojenliğini dikkate almaz ve özellikle yüzey işlemi görmüş altın takılar ve altın kaplamalı ürünler için doğru tespitler yapamaz. Yoğunluk yönteminin sınırlaması, alaşım türü yanlış değerlendirildiğinde önemli hatalara veya hatta yanlış sonuçlara yol açabilmesidir. Ancak, alaşım türü ve safsızlık elementlerinin göreceli oranları önceden biliniyorsa, ölçüm doğruluğu diğer yöntemlerinkinden daha üstündür. Bu nedenle, belirli uygulamalarda yoğunluk yöntemi ve X-ışını floresan spektroskopisinin birleştirilmesi, iki yöntemin doğrulama için birbirini tamamladığı çok etkili bir yaklaşımdır: Alaşım türünü tespit etmek için X-ışını floresan spektroskopisinin kullanılması, çeşitli safsızlık elementlerinin göreceli oranlarının kabaca ölçülmesi ve ardından içeriklerini belirlemek için yoğunluk yönteminin kullanılması, değerli metalin homojen bir alaşım olması ve altın kaplama veya altın dolgulu olmaması koşuluyla, mücevher kalite kontrol istasyonlarında yaygın olarak uygulanır.
4. X-ışını Floresan Spektrometresinin Nitel ve Nicel Analiz Yöntemleri
4.1 Numuneyi hazırlayın
Analizden önce numunenin çeşidi, izleri, görünümü vb. kontrol edilmeli; kirli yüzeyli numuneler, ölçüm yüzeyinin kirleticilerden arındırılmış olduğundan emin olmak için silinmelidir.
Kuyumculuk şirketleri, test kurumlarına ek olarak, üretim sırasında malzeme ve ürün kalitesini izlemek için X-ışını floresan spektrometrelerini yaygın olarak kullanırlar. Analiz edilecek numuneler katı veya sulu çözeltiler olabilir ve numunenin durumu ölçüm hatasını etkiler. Katı numuneler, kirleticilerden arındırılmış temiz yüzeylere sahip olmalıdır. Katı değerli metal numuneleri için, bileşen ayrışmasından kaynaklanan hatalara dikkat edilmelidir. Örneğin, ayrışma nedeniyle, aynı altın ağacından yapılmış ancak farklı konumlarda bulunan mücevher dökümleri farklı niteliklere sahip olabilir. Aynı kimyasal bileşime sahip ancak ısıl işlem görmüş numuneler farklı sayım oranları verecektir. Düzgün olmayan değerli metal numuneleri, düzgünlük sağlamak için yeniden eritilmeli, hızla soğutulmalı ve ardından levhalar haline getirilmeli veya kırılma yerlerinden alınmalıdır; düzgün olmayan yüzeyli numuneler düz bir şekilde cilalanmalıdır; toz numuneler için, 300-400 mesh'e öğütülmeli ve ardından disklere preslenmeli veya ölçüm için numune tutuculara yerleştirilmelidir. Sıvı numuneler filtre kağıdına damlatılarak, kızılötesi lamba ile nemleri kurutulduktan sonra ölçülebilir veya numune tutuculara kapatılabilir.
4.2 Numunenin ana elementlerini ve safsızlık bileşenlerini belirlemek için nitel analiz
Farklı elementlerin floresan X-ışınlarının belirli dalga boyları veya enerjileri vardır, bu nedenle elementlerin bileşimi floresan X-ışınlarının dalga boyuna veya enerjisine göre belirlenebilir. Dalga boyu dağılımlı bir spektrometre ise, X-ışınlarının dalga boyu λ, belirli bir düzlemler arası aralığa sahip bir kristal için dedektörün döndüğü 2ϴ açısından belirlenebilir ve böylece element bileşimi belirlenebilir. Enerji dağılımlı spektrometreler için enerji kanallar tarafından tanımlanabilir ve böylece hangi elementlerin ve bileşenlerin mevcut olduğu belirlenebilir. Ancak, element içeriği çok düşükse veya elementler arasında spektral çizgi girişimi varsa, manuel tanımlama yine de gereklidir. İlk olarak, X-ışını tüpünün hedef malzemesini belirleyin. X-ışınını ve eşlik eden güçlü pik çizgilerini ölçün, ardından kalan spektral çizgileri enerjiye göre etiketleyin. Bilinmeyen spektral çizgileri analiz ederken, kapsamlı bir karar vermek için kaynak ve numunenin özellikleri gibi faktörler dikkate alınmalıdır.
4.3 Standart numuneleri seçin ve kalibrasyon eğrilerini çizin
Nitel analiz sonuçlarına dayanarak, saflık seviyesi ve safsızlık bileşenleriyle eşleşen standart numuneler seçin. Genel olarak aşağıdaki gereklilikler geçerlidir:
(1) Standart numunedeki elementlerin türleri bilinmeyen numunedekilere benzer olmalı ve aynı olmalıdır.
(2) Standart numunedeki tüm bileşenlerin içeriği bilinmelidir.
(3) Standart numunedeki ölçülen elementlerin içerik aralığı, bilinmeyen numunedeki tüm ölçülen elementleri içermelidir.
(4) Standart numunenin durumu (toz numunelerin parçacık boyutu, katı numunelerin yüzey düzgünlüğü ve ölçülen elementlerin kimyasal durumu vb.) bilinmeyen numuneyle tutarlı olmalı veya uygun yöntemlerle tutarlı olacak şekilde işlenebilmelidir.
Her biri en az üç kez ölçülen numunelerin test edilmesi. Tekrarlanan ölçümlerden sonra, ortalama değer hesaplanmalı ve ardından her bir elementin içeriğinin standart değerleri ve ilgili ortalama değerler, kalibrasyon eğrisini çizmek ve doğrusal denklemi türetmek için parametre olarak kullanılmalıdır. Genellikle laboratuvarlar, kalibrasyon eğrisini düzenli olarak doğrulamalıdır.
4.4 Örnekleri tespit edin ve kantitatif analiz sonuçlarını hesaplayın
Örnek bir numune odasında test edilir ve bir elementin floresan X-ışını yoğunluğunun X-ışını floresan yoğunluğuna dayandığı gerçeğine dayanan X-ışını floresan spektrometrisi ile kantitatif olarak analiz edilir.Ben , C örneğindeki o elementin miktarıyla doğru orantılıdırBen
IBen = BenS x CBen
Formülde, benS C olduğunda elementin floresan X-ışını yoğunluğu nedir?Ben =100%.
Yukarıdaki formüle göre, kantitatif analiz standart eğri yöntemleri, artımlı yöntemler, dahili standart yöntemler vb. kullanılarak gerçekleştirilebilir. Ancak, bu yöntemler standart numunenin bileşiminin test numunesinin bileşimine mümkün olduğunca benzer olmasını gerektirir; aksi takdirde, test numunesinin matris etkisi, numunenin temel kimyasal bileşimindeki ve fiziksel ve kimyasal durumundaki değişiklikleri ifade eder ve bu da X-ışını floresansının yoğunluğunu etkiler. Kimyasal bileşimdeki değişiklikler, numunenin birincil X-ışınlarını ve X-ışını floresansını emmesini etkileyebilir ve ayrıca floresans artırma etkisini değiştirebilir.
Kalibrasyon eğrisine dayanarak, ölçülen değerleri kalibrasyon eğrisinin doğrusal denklemine yerleştirerek numune ölçümünün düzeltilmiş değerini hesaplayın. Her numune için, farklı konumlardan en az üç temsili test değeri seçin ve tekrarlanan ölçümler yoluyla bunların ortalamasını hesaplayın.
5. XRF Yönteminin Tespit Doğruluğunu Etkileyen Faktörler
XRF, benzer özelliklere sahip birçok standart maddedeki elementlerin floresan yoğunluğu ile içerikleri arasındaki ilişkiyi kullanarak matematiksel bir kalibrasyon eğrisi oluşturur ve ardından bilinmeyen numunelerdeki elementlerin floresan yoğunluğunu ölçerek içeriği belirler. Yüksek doğrulukta tespit sonuçları elde etmek için standart çalışma eğrisinin oluşturulması ve hesaplama yöntemlerinin seçilmesi çok önemlidir.
5.1 Standart Çalışma Eğrisi
Standart maddeler (standart numuneler), standart çalışma eğrilerinin oluşturulmasının temelini oluşturur. Ancak, şu anda iç piyasada değerli metal takılar için daha fazla ticari standart maddeye ihtiyaç duyulmaktadır ve değerli metal süs eşyalarındaki safsızlık türleri çeşitlidir. Safsızlık bileşimine uygun standart maddeler için gereklilikleri karşılamak zordur, çünkü bunlar yalnızca ticari olarak mevcut ulusal standart maddelere dayanmaktadır. Bu durum, matris etkileri nedeniyle analitik sonuçlarda önemli sapmalara yol açmaktadır. Örneğin, altın serisi standart maddelerinin kalibrasyonunda, nikel gibi safsızlık elementleri yoksa, nikel içeren beyaz K altını ölçmek için bir X-ışını floresan spektrometresi kullanmak kaçınılmaz olarak hatalara yol açacaktır.
Uyum için bir çalışma eğrisi oluştururken, düzeltme elemanlarını makul bir şekilde seçmek çok önemlidir. İster iyileştirme, ister emilim, ister örtüşme veya girişim olsun, eğri uydurma işleminden sonra hesaplanan hatalar ve standart numunelerin gerçek test sapmaları dikkate alınarak, seçilen elemanların ve yöntemlerin gerçekten etkili olup olmadığı belirlenmelidir.
Eğri uydurma sırasında en önemli kriter, eğri üzerindeki görünür içerik noktalarının önerilen değer noktalarına benzer olmasıdır. Hesaplanan düzeltme katsayıları, gerçek test sonuçlarının gerçek değerlerine daha yakın olabilmesi ve ölçüm verilerinin güvenilir ve doğru olması için pozitif ve negatif değerlere sahip olmalıdır.
5.2 Hesaplama Yöntemlerinin Seçimi
X-ışını floresan spektroskopisinde genellikle üç kantitatif analiz yöntemi kullanılır: doğrudan yöntem, fark yöntemi ve normalizasyon yöntemi.
(1) Doğrudan Yöntem. Au yoğunluğunu, ilgili yoğunluk ve içerik doğrusal ilişki denklemine yerleştirerek Au içeriğini hesaplar.
(2) Fark Yöntemi. Toplam 0 miktarından safsızlık elementlerinin içeriğinin doğrudan çıkarılmasıyla ana elementin içeriği elde edilir.
(3) Normalizasyon Yöntemi. Normalleştirilmiş içeriğin 0 olduğunu varsayar, her bir elemanın içerik değerlerini toplar ve 0 ile karşılaştırır. Fazla kısım, her bir elemanın nihai içerik değerlerini elde etmek için ağırlıklandırılır.
Test edilecek değerli metal elementinin içeriği 'ten fazla olduğunda, ana element içeriği ile yoğunluk arasındaki doğrusal ilişki zayıflar ve doğrudan doğrusal ilişkiden elde edilen sonuçlar genellikle hatalı olur. Safsızlık elementlerinin doğrusal ilişkisine geçmek, nispeten doğru safsızlık elementi içeriği sağlayabilir. Normalizasyon yöntemi veya fark çıkarma yöntemi kullanılarak daha doğru ana element içeriği elde edilebilir. Değerli metal elementlerinin içeriği 'ten az olduğunda, yoğunluk ile Au içeriği arasındaki doğrusal ilişkinin hesaplamalarda doğrudan kullanılması daha doğru sonuçlar verir.
Copywrite @ Sobling.Jewelry - Özel takı üreticisi, OEM ve ODM takı fabrikası
Bölüm V Ateş Analizi Yöntemi (Kupelasyon Yöntemi)
Kupelasyon yöntemi olarak da bilinen ateş analizi, eritme ve kavurma yoluyla mineraller ve metal ürünlerdeki değerli metal bileşenlerinin içeriğini belirler. Ateş analizi, yalnızca altın ve gümüşü zenginleştirmenin eski bir yolu değil, aynı zamanda altın ve gümüş analizi için de önemli bir yöntemdir. Yurt içinde ve yurt dışında jeoloji, madencilik ve altın ve gümüş eritme endüstrileri, bu yöntemi üretimde en güvenilir analitik yöntem olarak yaygın olarak kullanmaktadır.
Ateş analizi, uluslararası alanda en doğru yöntem olarak kabul edilmektedir. Birçok ülke bunu ulusal standart olarak belirlemiş ve altın içeriğini belirlemek için uluslararası alanda kabul görmüş bir tahkim yöntemi haline gelmiştir. Çin'in "Mücevherlerdeki Değerli Metallerin Saflığına İlişkin Düzenlemeler ve Adlandırma Yöntemleri" (GB 11887-2012) standardı da ateş analizini altın alaşımlarındaki altın içeriğini ölçmek için tahkim yöntemi olarak belirlemiştir.
1. Ateş Analizi Yönteminin Prensibi
Analiz edilecek altın numunesinden belirli bir kütle tartılır, uygun miktarda gümüş eklenir, kurşun folyoya sarılır ve yüksek sıcaklıkta eritilir. Erimiş kurşun, altın, gümüş ve değerli metalleri yakalayarak, erimiş haldeki açığa çıkan altın ve gümüşü tamamen çözer. Erimiş alaşımdaki kurşun, havada veya oksijende kolayca oksitlenerek erimiş kurşun oksit oluşturur. Kurşun oksidin yüzey gerilimi ve bağıl yoğunluğu erimiş kurşundan farklıdır ve erimiş kurşunun dibe çökmesine ve bir kurşun düğmesi oluşturmasına neden olur. Aynı zamanda, gözenekli kül tabağı, ıslatma özellikleri ve kılcal etkisi nedeniyle erimiş kurşun oksidi emer. Erimiş kurşunun kohezif kuvveti güçlüdür ve kül tabağı tarafından emilmez. Erimiş kurşun oksit kül tabağına sızdıktan sonra, erimiş kurşun yeni bir yüzey ortaya çıkarır ve tekrar oksitlenir ve kül tabağı yeni oluşan erimiş kurşun oksidi emer. Bu işlem, tüm kurşun oksitlenip kül tablası tarafından emilene kadar tekrar tekrar devam eder ve kurşun külçesi ile cürufun iyi bir şekilde ayrılması sağlanır. Bu işlem sırasında, diğer baz metal elementleri de kısmen veya tamamen uçucu oksitler oluşturabilir veya kül tablası tarafından emilerek, safsızlık elementlerinin uzaklaştırılması ve daha saf değerli metal parçacıkları elde edilmesi hedeflenir. Kül üfleme işleminden sonra, alaşım parçacıkları, gümüşün nitrik asitte çözünürken altının çözünmemesi özelliğinden yararlanılarak işlenir ve gümüş nitrik asitte çözülerek altın ayrıştırılır. Numunenin altın içeriği, nitrik asitten ayrıştırılan altın tartıldıktan ve aynı anda ölçülen saf bir altın standart numunesiyle düzeltildikten sonra hesaplanır.
2. Yangın Testinin Avantajları ve Dezavantajları
2.1 Avantajları
(1) Ateş testi yöntemi geniş bir uygulama alanına sahiptir ve 3,0 ile 9,5 arasında altın içeriğine sahip çeşitli altın ve K altın takılardaki altın içeriğini belirlemek için kullanılabilir. Mücevher endüstrisi test kurumlarında klasik bir test yöntemi olarak kabul edilmektedir.
(2) The analysis results are reliable, with high precision and accuracy.
(3) The sample size is large and representative, which can significantly reduce sampling errors.
2.2 Disadvantages
(1) It is a destructive method requiring the destruction of samples for testing, resulting in high detection costs.
(2) It is not suitable for samples of high-purity gold jewelry (gold content above 999.5%) and samples containing impurities that are insoluble in nitric acid (such as Ir, Pt, Rh, etc.).
(3) The ash fusion process requires using the harmful element Pb as a collector, posing safety risks to the health of inspectors and the environment.
(4) The analysis process is lengthy, with many experimental steps and complex operations, requiring a high level of professional skills and experience from the experimental personnel.
3. Equipment and utensils used in the fire assay method
3.1 Ash Blowout Furnace
The high-temperature Ash Blowout Furnace is used for fire assay (a muffle furnace). The muffle furnace specifically designed for ash blowing should have air intake and exhaust ports to allow for air circulation, preferably capable of preheating the air and ensuring stable passage, as shown in Figure 6-5, with the furnace temperature able to be evenly heated from room temperature to 1100℃.

3.2 Analytical Balance
The fire assay method is a quality analysis method that has strict requirements for the analytical balance, generally requiring a sensitivity of precision analytical balance within 0.01mg. The balance and weights must be calibrated regularly, with calibration cycles ideally set to 1 month or one quarter, depending on the workload.
3.3 Gold Separation Basket
The materials used to make gold separation baskets vary by country. In China, assay laboratories often use platinum or stainless steel plates, as shown in Figure 6-6.

3.4 Rolling Mill
Used to compress the alloy into thin sheets, requiring the thickness of the rolled sheets to be uniform and consistent to avoid increasing analytical errors.
3.5 Ash Dish
The ash dish is a porous refractory vessel that absorbs lead oxide during the lead-blowing process. Common ash dishes include cement ash dishes, bone ash cement ash dishes, and magnesia ash dishes (Figure 6-7).

4. Analysis Steps of Fire Assay
Taking gold alloy jewelry with gold content between 333.0% and 999.5% as an example, the process of analyzing its gold content is mainly divided into eight steps: pre-analysis, weighing, silver replenishment, lead cladding, ash blowing, rolling, gold separation and calculation of results.
4.1 Pre-analysis
Common pre-analysis methods include the weight method and X-ray fluorescence spectroscopy (XRF). The weight method has higher accuracy for pre-analysis but takes longer. The XRF method is fast and can simultaneously analyze the impurity element content in the sample, but it has a larger margin of error. For general samples, XRF can be used for pre-analysis to understand the basic composition of the sample, facilitating the calculation of the quality of standard samples of silver, copper, nickel, etc. For irregular shapes or samples with larger XRF analysis errors, the weight method can be used for pre-analysis.
4.2 Weighing
Weigh 200-300mg standard gold samples in three or four portions and three or four portions of test samples equivalent to the standard gold quality, accurate to 0.01mg. The samples should be cut into small pieces, mixed evenly, and weighed to make the weighing more representative. The weighing of standard gold and samples should follow the principle of consistency, with the component ratios as similar as possible. The weighing deviation between parallel standard gold and parallel samples should be controlled within 2%.
4.3 Silver replenishment
When supplementing silver, the ratio of silver to gold is crucial. If silver is less than twice the amount of gold, the gold separation cannot proceed. A large ratio of gold to silver can easily cause the gold roll to break. It is more appropriate for the amount of silver to be 2.1-2.5 times that of gold. The extremely poor silver amount should be controlled within 1%. Considering the total amount of base metals contained in the sample, an appropriate amount of copper should be added proportionally to the standard gold.
4.4 Lead cladding
Wrap the weighed standard gold and sample separately in lead foil, roll them up, and number them. The weight of the lead foil is generally 3.5g, and the lead packaging amount for the standard gold and sample should be as consistent as possible. The amount of lead is proportional to the impurity content of the sample; if the copper and nickel content is high, the amount of lead can be increased. The lead and sample should be tightly wrapped to minimize gaps, avoiding splashing losses caused by air expansion after the lead is placed, as shown in Figure 6-8.
Note: The numbers in the figure are sample numbers; the same applies below
4.5 Ash blowing
Place the standard gold wrapped in lead foil and the sample into the ash-blowing furnace, arranging the standard gold and sample in a cross pattern to avoid temperature discrepancies. The crucible should be preheated to above 920℃ to prevent residual organic matter and other volatiles from causing splattering. Maintain the furnace temperature at 920-1000℃, and continue heating in an oxidizing atmosphere until the sample completely melts, for about 25 minutes. If using a closed ash-blowing furnace, after maintaining it at 920-1000℃ for 30-40 min, slightly open the furnace door for oxidizing ash blowing, and close it after 10-15 min.
After ash blowing is complete, stop heating and allow the furnace to cool down to below 700℃ before removing it, as shown in Figure 6-9, to avoid rapid cooling that could cause rapid oxidation of the agglomerates, leading to splattering and spiking.

4.6 Rolling
Use a brush to remove the ash materials adhered to the alloy particles, flatten them on an anvil (Figure 6-10), and then anneal at 700℃. Use a rolling mill to roll the alloy particles into thin sheets of 0.15-0.2 mm (Figure 6-11), and then anneal again, avoiding excessive time. The direction in which the alloy particles are fed during rolling should be consistent to prevent sample cracking and loss. The thickness of the rolled sheets should be uniform to ensure consistency in value addition. Use a digital steel stamp to mark and roll into a cylindrical shape (Figure 6-12).
Figure 6-10 Flattening the alloy particles

Figure 6-11 Rolling thin sheets

4.7 Gold Separation
Use nitric acid to dissolve silver from the gold alloy roll. Before separating the gold, clean the alloy roll, flask, or basket to prevent contamination or the introduction of chloride ions. Immerse the gold roll in a separation flask containing 20mL of near-boiling nitric acid, keeping it always below the boiling point at a temperature close to boiling, and heat continuously for 15 minutes or until the nitrogen oxide salt mist is driven off, as shown in Figure 6-13. Slowly pour out the solution, wash the gold roll with hot water 3-5 times, and then immerse it in boiling nitric acid and wash it again.

Carefully transfer the standard gold after separation to a porcelain crucible, dry it, and burn it to a golden yellow, as shown in Figure 6-14. After cooling, weigh the mass of the gold roll, which is accurate at 0.01mg.
4.8 Calculation result
Gold content Wt(Au) is calculated according to formula (6-3), with the result rounded to one decimal place:
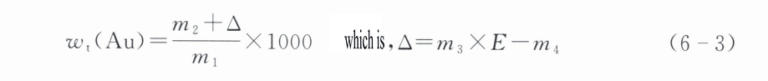
Formülde:
m1 – sample mass (g);
m2 – a mass of the gold obtained after separating from the sample (g);
m3 – a mass of standard gold (g);
m4 – The quality (g) of the gold ingot obtained after standard gold analysis;
E – The purity of standard gold (‰).
The deviation in results caused by repeated experiments should be less than 0.2‰ for 999.0‰-999.5‰; Gold alloys should be less than 999.0‰ and less than 0.5‰; white K gold should be less than 1‰.
5. Factors Affecting the Accuracy of Fire Assay Analysis
When applying fire assay to analyze gold content, factors such as sample size, type of cupel furnace, material of the cupel, silver to gold ratio, cupellation temperature, and separation time will all affect the results. It is necessary to conduct accompanying experiments using gold standard samples and maintain consistency in the analysis conditions of the gold standard samples and the samples to obtain good parallelism and accurate, reliable results, eliminating systematic errors in the analysis process.
5.1 Sample Size
The sample size for analyzing K gold jewelry is generally small, related to the high content of alloying elements in K gold jewelry. However, a sample size that is too small will directly affect the sample’s representativeness and the analysis’s accuracy. The sample size can be appropriately increased for jewelry with higher purity and lower nickel and copper content for better results. For lower-grade K gold, the amount of lead foil can be appropriately increased to facilitate the separation of impurities. The standard gold appreciation should have a certain range of control and trade-offs to avoid systematic deviations.
5.2 Ash blowing Furnace
A regular muffle furnace can only meet the temperature requirements. It cannot provide the oxidizing gas flow needed during the cupellation process, which reduces the quality and effectiveness of the cupellation. Additionally, it poses certain safety hazards: to provide the oxygen required for oxidation, the furnace door must be opened slightly during the cupellation stage, causing a large amount of lead oxide to escape from the furnace door, resulting in serious lead contamination of the surrounding environment and endangering the health of the operators. Furthermore, prolonged use can lead to corrosion damage of the furnace chamber and opening by lead oxide, and the large amount of lead residue inside the furnace is difficult to discharge in time, which can easily contaminate the analysis samples. Therefore, a dedicated cupellation furnace should be prioritized.
5.3 Ashtray Material
When selecting the material and ratio for ashtrays, it is important to consider not only the ashtray’s ability to absorb impurity elements in the lead but also the recovery rate of gold and silver during the ash-blowing process. Magnesia ashtrays have a relatively high recovery rate, but there are issues with removing adhered particles at the bottom and determining the ash-blowing temperature and endpoint. Bone ash and cement ashtrays make it easier to judge and control the ash-blowing temperature and endpoint, resulting in purer aggregates that are less likely to break when struck into thin sheets. However, the recovery rate is relatively lower.
5.4 Silver to Gold Ratio
Silver has two roles in fire assay: extraction, which extracts gold from impurities, and protection, which reduces gold loss during the measurement process. A small amount of silver can increase gold loss and incomplete oxidation during ash blowing, but more silver is not always better. When the amount of silver added is three times the weight of gold, gold loss increases, and the gold can easily break during separation. Generally, the amount of silver added is related to the sample’s composition. During ash blowing, when nickel and palladium in white K gold alloys are captured, gold can also be lost, so a larger amount of silver is usually required as a protective agent to prevent gold loss. When analyzing gold content using fire assay for white gold alloys containing nickel but not palladium, nickel should be added to the standard gold in quantity roughly equivalent to the sample, and the amount of lead should be increased. For white gold alloys containing palladium, palladium should be added to the standard gold in a quantity roughly equivalent to the sample while increasing the lead amount.
5.5 Ash Blowing Temperature
Taking 18K gold as an example, under the same process conditions, when the ash-blowing temperature is within the 900-1500℃ range, the standard gold loss increases with the rise in ash-blowing temperature and shows a linear distribution. When the ash-blowing temperature is too high, silver is prone to evaporation and splashing, leading to increased errors in the analysis results; when the ash-blowing temperature is too low, the molten lead oxide and impurities may also clump together, which cannot be fully absorbed by the crucible, resulting in the analysis process being unable to proceed.
5.6 Gold Separation Time
Taking 18K white gold as an example, the gold measurement results decrease as the gold separation time increases, but after reaching a certain level, the gold measurement results remain unchanged.
Section VI Inductively Coupled Plasma Emission Spectroscopy (ICP Method)
The inductively coupled plasma emission spectrometer, also known as the ICP spectrometer or ICP atomic emission spectrometer, uses inductively coupled high-frequency plasma as the excitation light source, utilizing the characteristic emission spectra of each element’s atoms or ions to determine the composition of substances and conduct qualitative and quantitative analysis of elements. ICP discharge is a relatively simple and highly effective method that transforms aerosols and vapors of liquids and solids, as well as gases at normal pressure, into free atoms, excited state atoms, and ions or molecular fragments. It can quickly analyze various major, trace, and ultra-trace elements in materials. It is one of the most competitive methods for simultaneous multi-element analysis, characterized by a wide testing range, fast analysis speed, and low detection limits. It has high precision and accuracy for detecting high-content gold and is a commonly used method by jewelry industry testing institutions for determining high-content gold jewelry materials.
1. Principle of the ICP Method
The working principle of the ICP method is shown in Figure 6-15.
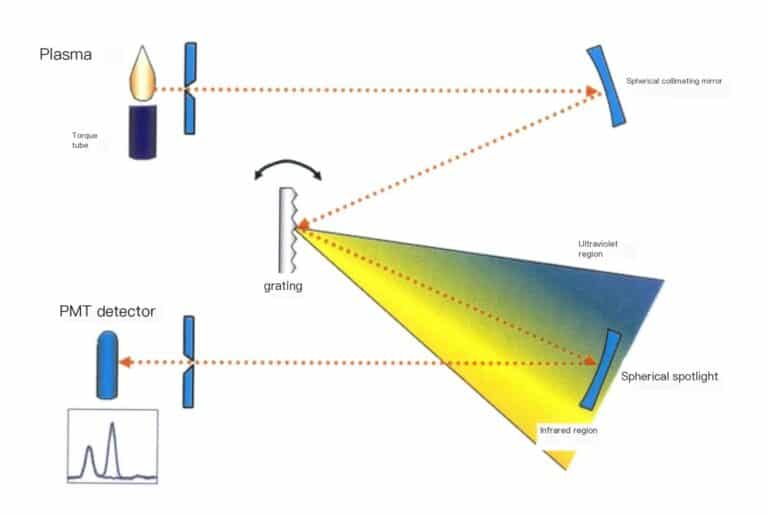
The high-frequency power generated by the radio frequency generator is applied to the three-layer concentric quartz torch tube through the induction working coil, forming a high-frequency oscillating electromagnetic field; argon gas is introduced into the outer layer of the quartz torch tube, and high-voltage discharge is performed to generate charged particles. The charged particles move back and forth in the high-frequency electromagnetic field, colliding with other argon ions, producing more charged particles. At the same time, the temperature rises ultimately forming argon plasma, with temperatures reaching 6000-8000 K. The sample of the aqueous solution to be tested is passed through the atomizer. The formed aerosol enters the central channel of the quartz torch tube, where it is fully evaporated, atomized, and ionized in high temperature and inert gas, emitting characteristic spectral lines of the elements contained in the solution; by collecting light from the plasma light source and using a scanning spectrometer for scanning, the light intensity of the characteristic spectral lines of the elements to be tested is accurately positioned at the exit slit. The light intensity of the spectral line is converted into photoelectric current using a photomultiplier tube. After circuit processing and analog-to-digital conversion, it enters the computer for data processing. The presence or absence of characteristic spectral lines identifies whether a certain element is present in the sample (qualitative analysis); based on the intensity of the characteristic spectral lines determines the content of the corresponding element in the sample (quantitative analysis).
2. Advantages and Disadvantages of the ICP Method
2.1 Avantajları
(1) Ability to detect multiple elements simultaneously. It can detect multiple elements in the same sample at the same time. Once the sample is excited, each element emits its characteristic spectral lines, allowing for separate detection while simultaneously determining multiple elements.
(2) Fast analysis speed. Most samples can be analyzed without chemical treatment, and solid and liquid samples can be analyzed directly. Additionally, multiple elements can be determined simultaneously. Using a photonic direct-reading spectrometer, the quantitative determination of dozens of elements can be completed within minutes.
(3) Good selectivity. Due to the strong characteristic of the spectrum, it is particularly significant for analyzing elements with very similar chemical properties. For example, analyzing dozens of rare earth elements in Nb and Ta, Zr, and Hf is very difficult compared to other methods. At the same time, emission spectroscopy can easily distinguish and measure them.
(4) Low detection limit. The detection limit of a general light source is (0.1-10) x 10-6, with an absolute value of (0.01-1) 10-6; while using an inductively coupled plasma (ICP) light source, the detection limit can be as low as 10-9 orders of magnitude.
(5) Higher accuracy. The relative error of a general light source is 5% to 10%, while the relative error of ICP can reach below 1%.
(6) The linear range of the ICP light source standard curve is wide, reaching 46 orders of magnitude, allowing for multi-element analysis of a single sample, and can measure different concentrations of high, medium, and low levels.
(7) Low sample consumption, suitable for the multi-component determination of whole batches of samples, especially qualitative analysis, shows unique advantages.
2.2 Disadvantages
The disadvantages of the ICP method are as follows.
(1) Many factors affect the intensity of the spectral lines, such as sample composition, uniformity, sample parallelism, acid concentration, spectral interference, temperature, and humidity, all of which can impact the final detection results. There are high requirements for the components of the standard reference, and most non-metallic elements have difficulty obtaining sensitive spectral lines.
(2) Solid samples generally need to be converted into a solution beforehand, which often worsens the detection limit; the accuracy is poor when the concentration is high.
(3) Not suitable for samples containing impurities such as Ir that are insoluble in aqua regia.
(4) Requires an expensive inductively coupled plasma emission spectrometer, which consumes a large amount of argon during operation, resulting in high detection costs.
3. Instruments and reagents used in the ICP method
3.1 Instruments
Instruments include: Inductively coupled plasma emission spectrometers、Beakers、Volumetric flasks and so on other common laboratory glassware, high-precision electronic balances and etc.
3.2 Reagents
The water used for ICP testing meets the specifications for first-grade water or water of equivalent purity as specified in “Specifications and Test Methods for Water Used in Analytical Laboratories” (GB/T 6682-2008).
The chemical reagents used in ICP testing can be divided into two categories: sample decomposition and preparing standard solutions of elements. All reagents are required to be of analytical grade. When analyzing gold content, a high-purity gold sample with a purity not lower than 99.999% is needed.
4. ICP Analysis Steps
As an example, the steps include the following analysis of gold content in gold jewelry.
4.1 Sample Preparation
After grinding the sample thin, Cut it into small pieces, place them in a beaker, add 20mL of ethanol solution, heat, boil for 5 minutes, then remove it. Pour off the ethanol solution, and wash the gold piece repeatedly with ultrapure water three times. Add 20mL of hydrochloric acid solution, heat, boil for 5 minutes, then remove it. Pour off the hydrochloric acid solution, and wash the gold piece repeatedly with ultrapure water three times. Place the gold piece in a glass weighing bottle, cover it, and put it in the oven to dry at 105℃, then take it out for later use.
4.2 Solution Preparation
(1) Sample Solution. Weigh (1000±2.5)mg sample (accurate to 0.01mg), place it in a 100mL beaker, add 30mL of aqua regia, cover with a watch glass, and slowly heat until completely dissolved, continuing to heat to remove nitrogen oxides. After cooling, transfer the solution to a 50mL volumetric flask, rinse the watch glass and beaker with aqua regia solution, add the wash liquid to the volumetric flask, dilute to the mark, and mix well for later use. Prepare two portions of sample solution for each sample.
(2) Calibration Solution. Weigh three portions of high-purity gold samples with a mass of (1000±2.5)mg (purity > 99.999%), dissolve them to obtain three portions of high-purity gold solution, and prepare the calibration solution according to the following steps.
Calibration Solution 1:
Transfer the first portion of the high-purity gold solution to a 50mL volumetric flask. Rinse the watch glass and beaker with aqua regia solution. Add the wash liquid to the volumetric flask, Dilute to the mark, Mix well. The concentration of the measured impurity elements in Calibration Solution 1 is set to 0/ug/m.
Calibration solution 2:
Transfer the second portion of the high-purity gold solution to a 50mL volumetric flask pre-filled with 5mL of mixed standard solution 1. Rinse the surface dish and beaker with aqua regia solution. Add the wash liquid to the volumetric flask. Dilute to the mark. Shake well.
Calibration solution 3:
Transfer the third portion of the high-purity gold solution to a 50mL volumetric flask pre-filled with 5mL of mixed standard solution 2. Rinse the surface dish and beaker with aqua regia solution. Add the wash liquid to the volumetric flask, Dilute to the mark, Shake well.
4.3 Determination
Adjust the ICP spectrometer to optimal conditions; if testing gold alloy samples, select appropriate analytical lines and background correction according to Table 6-2.
Table 6-2 Recommended wavelengths for impurity elements (analytical lines) (Unit: nm)
| Element | Wavelength | Other available wavelengths | Element | Wavelength | Other available wavelengths |
|---|---|---|---|---|---|
| Ag | 328.068 | 338.289 | Ni | 352.454 | 231.604 |
| Al | 396.152 | 308.215 | Pb | 168.220 | 220.353 |
| As | 189.042 | 193.696 | Pd | 340.458 | 355.308 |
| Bi | 223.061 | 306.772 | Pt | 306.471 | 203.646 |
| Cd | 226.502 | 228.802 | Rh | 343.489 | - |
| Co | 228.616 | 238.892 | Ru | 240.272 | - |
| Cr | 267.716 | 283.563 | Sb | 206.833 | 217.581 |
| Cu | 324.754 | 327.396 | Se | 196.090 | - |
| Fe | 259.940 | 239.563 | Sn | 189.989 | 189.927 |
| Ir | 215.278 | - | Te | 214.281 | - |
| Mg | 279.553 | 280.270 | Ti | 334.941 | - |
| Mn | 257.610 | 260.569 | Zn | 213. 856 | - |
Measure the impurity element spectral line intensity of calibration solution 1,3, where the concentration of the measured impurity elements in calibration solution one is set to 0/ug/mL, and plot the working curve based on the test results; under the same conditions as the measurement calibration solution, measure the spectral line intensity of impurity elements in two sample solutions and obtain the concentration of each impurity element in the sample solution from the working curve.
4.4 Result Representation
(1) Calculation of the total amount of impurity elements. The total amount of impurity elements in the sample is calculated according to formula (6-4):

Formülde:
ƩA – total amount of impurity elements in the sample (‰);
ƩCBen – total concentration of impurity elements in the sample solution ug/mL);
V – volume of the sample solution (mL);
m – Mass of the sample (mg).
(2) Calculation of gold content.
The gold content in the sample is calculated according to formula(6-5):

Formülde:
w(Au) – gold content in the sample (‰);
ƩA – Total amount of impurity elements in the sample (‰).
(3) Reproducibility. The relative deviation of the total impurity elements in two parallel determinations of the samples should be less than 20%; if exceeded, re-determination is required.
5. Factors of Interference in ICP Analysis
During the ICP detection process, interference phenomena are inevitably present, as shown in Figure 6-16. Based on the interference mechanism, it can be divided into two main categories: spectral interference and non-spectral interference. In contrast, according to the state of the interfering factors, it can be divided into gas-phase interference and condensed-phase interference.
Spectral interference and non-spectral interference are effects caused by the components of the sample matrix and accompanying substances, which enhance or weaken the already resolved analytical signals. Non-spectral interference includes sample preparation, spray, solvent removal, volatilization, atomization, excitation, and ionization interference, as shown in Figure 6-16.
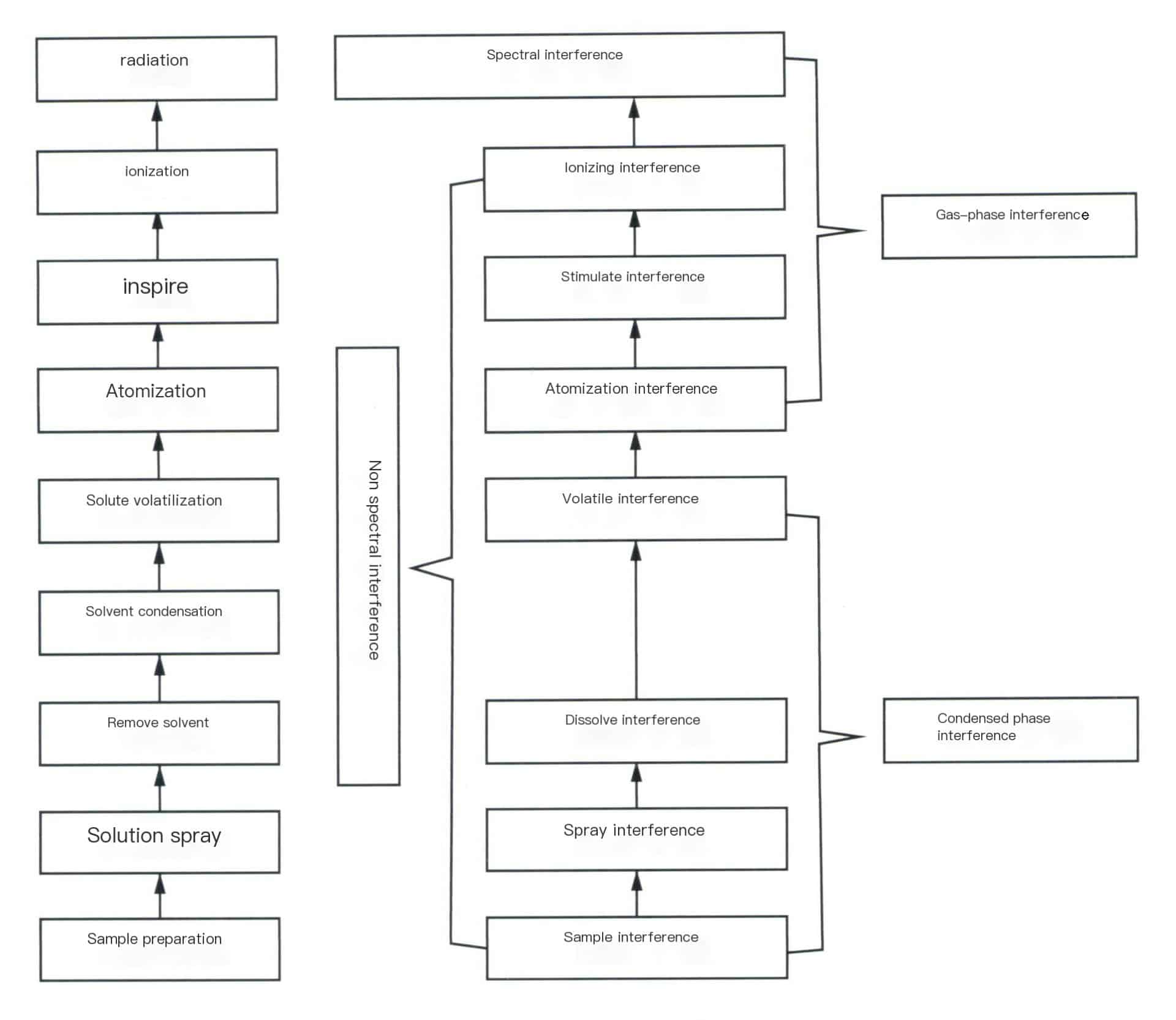
5.1 Spectral Interference
Spectral interference is caused by the inability to resolve the radiation signals generated by the analyte signals and interfering substances. It is the most important and troublesome issue in ICP spectrometry. Due to the strong excitation capability of ICP, almost every substance present in or introduced into ICP emits a considerable number of spectral lines, resulting in a large amount of spectral “interference.”
Spectral interference is mainly divided into two categories: one is line overlap interference, which is caused by the insufficient dispersion and resolution of the spectrometer, leading to the overlap of spectral lines of certain coexisting elements in the analysis; the other is background interference, which is related to the influence of the matrix composition and the strong stray light emitted by the ICP light source itself. Using a high-resolution spectroscopic system for line overlap interference does not mean that this type of spectral interference can be eliminated; it can only be considered that when spectral interference occurs, it can be reduced to minimum intensity. Therefore, the most commonly used method is to select another spectral line with less interference as the analytical line or to apply interference factor correction (IEC) for correction. The most effective method for background interference is to utilize the background correction technology available in modern instruments to deduct it. When interference occurs, they can be reduced to a minimum intensity. Therefore, the most commonly used method is to select another spectral line with less interference as the analytical line or to apply interference factor correction (IEC) for correction. The most effective method for background interference is to utilize the background correction technology available in modern instruments to deduct it.
5.2 Non-spectral interference
(1) Interference from physical factors.
Since the samples for ICP spectral analysis are in solution form, factors such as the viscosity, relative density, and surface tension of the solution all affect the atomization process, droplet size, aerosol transport, and solvent evaporation, and viscosity is related to the composition of the solution, the concentration and type of acid, and temperature.
When the solution contains organic solvents, both dynamic viscosity and surface tension will decrease, improving atomization efficiency. At the same time, most organic reagents are flammable, thus increasing the tail flame’s temperature and increasing spectral line intensity. At this time, the power of the ICP needs to be appropriately increased to suppress the intensity of the molecular spectrum of carbides in the organic reagents.
As seen above, the interference of physical factors exists and should be avoided. The main method is to ensure that the standard test solution and the sample to be tested are completely consistent regarding the composition of matrix elements, total salinity, concentration of organic solvents, and acids. Currently, the peristaltic pump sampling system used can help reduce the aforementioned physical interference. Additionally, internal standard correction can appropriately compensate for the effects of physical interference. Matrix matching or standard addition methods can effectively eliminate physical interference but require a larger workload.
(2) Ionization interference.
Since the sample in the ICP is evaporated, dissociated, ionized, and excited in the channel, the changes in sample composition have little effect on the electrical parameters of the high-frequency skin effect. Therefore, the influence of easily ionizable elements on the intensity of ion lines and atomic lines is smaller than that of other light sources. However, experiments show that this easily ionizable interference effect still has a certain impact on spectral analysis.
For vertically observed ICP light sources, appropriately selecting the plasma parameters can minimize ionization interference.
However, for horizontally observed ICP light sources, this easily ionizable interference is relatively more severe. The currently used bidirectional observation technology can effectively address this easily ionizable interference. It is also necessary to maintain a similar composition between the sample solution to be tested and the analytical standard solution.
(3) Matrix effect interference.
The matrix effect originates from the plasma, and for any analytical line, this effect is related to the excitation potential of the spectral line. However, due to the good detection capability of ICP, the analytical solution can be appropriately diluted to keep the total salt content around 1mg/mL. In this diluted solution, matrix interference is often negligible. When the concentration of the matrix substance reaches several milligrams per milliliter, the matrix effect cannot be completely ignored. The matrix effect of the ICP light source is slightly more severe when observed horizontally. Matrix matching, separation techniques, or standard addition methods can eliminate or suppress the matrix effect.
Copywrite @ Sobling.Jewelry - Özel takı üreticisi, OEM ve ODM takı fabrikası